La deficit de α-N-acetilgalactosaminidază este o boală de depozitare lizozomală care apare foarte rar. Este împărțit într-o formă juvenilă și adultă.
Care este deficitul de a-N-acetilgalactosaminidază?

Deficitul de a-N-acetilgalactosaminidază sau Deficitul de Alpha-N-acetilgalactosaminidază se mai numește și în medicină Boala Schindler, Boala Schindler sau Boala Kanzaki desemnat. Ceea ce se înțelege este o boală lizozomică extrem de rară, a cărei moștenire este recesivă autosomală. Medicii diferențiază între o formă juvenilă, care apare la copii și adolescenți, și o formă adultă, de care suferă adulții.
Forma juvenilă a deficienței de alfa-N-acetilgalactosaminidază se numește boala Schindler, în timp ce forma adultă se numește boala Kanzaki. Există, de asemenea, subdiviziuni în boala Schindler tip I pentru forma tinerească și boala Schindler tip II pentru forma adultă. Un amestec de ambele forme, cunoscut sub numele de boala Schindler tip III, este de asemenea posibil.
Toate cele trei forme ale bolii sunt oligozaharidozele (glicoproteinozele), care includ fucozidoza și boala de stocare a acidului sialic. Denumirea boala Schindler sau boala Schindler pentru deficiența de alfa-N-acetilgalactosaminidază se întoarce la geneticianul german Detlev Schindler.
El a fost primul care a descris boala în 1988. Un an mai târziu, medicul japonez T. Kanzaki a descris forma adultă. El a descoperit în 1991 că o deficiență de alfa-N-acetilgalactosaminidază a fost cauza bolii.
cauze
Deficitul de α-N-acetilgalactosaminidază este cauzat de o activitate redusă a enzimei alfa-N-acetilgalactosaminidază. Misensiunea sau mutațiile prostii ale genei NAGA, care codifică alfa-N-acetilgalactosaminidaza, sunt responsabile pentru defectul enzimei.
Gena NAGA este localizată pe gena locos q11 a cromozomului 22. Enzima alfa-N-acetilgalactosaminidază are sarcina de a cataliza clivajul de N-acetilgalactosamină din diferite glicolipide și glicoproteine. Defectul genetic determină o activitate redusă a enzimei, astfel încât substanțele care nu sunt metabolizate se acumulează în celulele corpului.
În cele mai multe cazuri, acestea sunt proteoglicane, glicosfingolipide și glicoproteine legate de N sau O. Acumularea acestora provoacă daune celulei și organelor afectate. Deficitul de Alpha-N-acetilgalactosaminidază este atât de rar încât nu există informații precise despre frecvența sa.
Până în prezent, doar doisprezece pacienți din opt familii sunt cunoscuți că au dezvoltat boala Schindler în lume. Plângerile neurologice nu apar la fiecare pacient. Unii medici suspectează influența unor factori suplimentari pentru apariția simptomelor neurologice. Cu toate acestea, încă nu există dovezi care să susțină această presupunere.
Vă puteți găsi medicamentul aici
➔ Medicamente pentru durereSimptome, afectiuni si semne
Simptomele deficitului de alfa-N-acetilgalactosaminidază depind de forma particulară a bolii. În contextul bolii lui Schindler de tip I, hipotonia musculară progresivă apare în primul an de viață. În plus, bebelușii afectați suferă de o regresie psihomotorie accentuată.
Au fost de asemenea înregistrate ca simptome tulburări ale secvenței de mișcare, tetraplegia spastică și convulsii cerebrale mioclonice. De asemenea, există riscul ca copilul să devină orb. Dacă există o formă adultă de deficiență de alfa-N-acetilgalactosaminidază, adică boala Schindler tip 2, apar simptome similare cu boala Fabry.
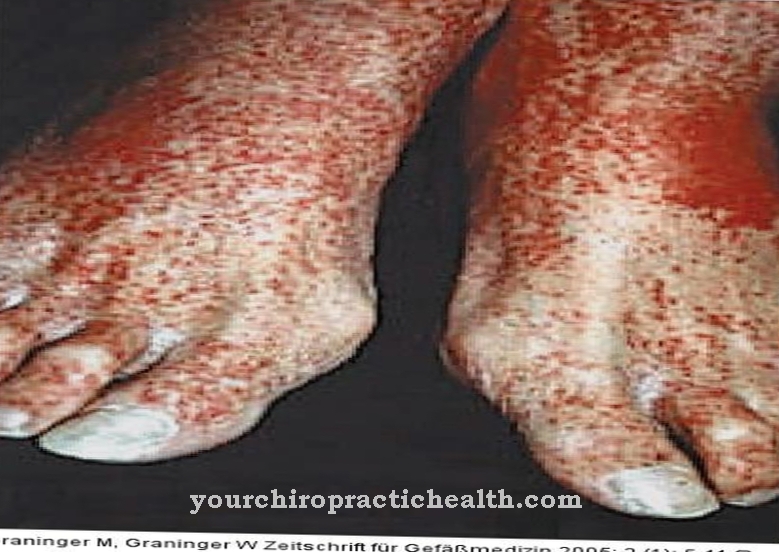
Pacienții au angiokeratoame, care sunt modificări benigne ale pielii. Mulți pacienți sunt ușor cu retard mental. O formă intermediară clinică este boala Schindler de tip III. Cei afectați suferă de disfuncții neurologice, convulsii cerebrale și dizabilități mintale.
Cu toate acestea, sunt posibile forme mai puțin severe, care sunt asociate cu plângeri psihice și neurologice ușoare, dezvoltarea întârziată a limbajului sau simptome similare cu autismul.
Diagnostic și curs
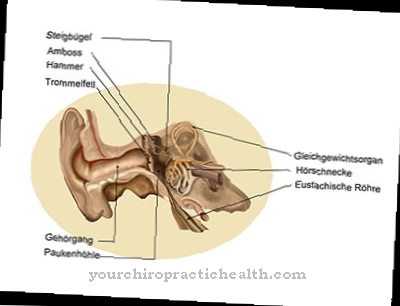
Diagnosticul unei deficiențe de alfa-N-acetilgalactosaminidază este posibil prin detectarea unei activități reduse de NAGA. În acest scop, testele enzimatice sunt efectuate în plasma de sânge, leucocite sau fibroblaste sau limfoblaste de cultură. Detectarea cromatografică a oligozaharidelor în urină permite, de asemenea, diagnosticul deficitului enzimatic.
O analiză ADN poate fi, de asemenea, efectuată, dar acest lucru nu este necesar în majoritatea cazurilor. În timpul sarcinii, diagnosticul prenatal al deficienței de enzimă poate fi, de asemenea, efectuat folosind o analiză mutațională a genei NAGA în urma unei prelevări de coriuni de vilozitate sau amniocenteză. Cu toate acestea, forma respectivă a bolii nu poate fi determinată.
Diagnosticul diferențial al sindromului Fabry, neurodegenerarea asociată pantofenatului kinazei și distrofiei neuroaxonale infantile, care au și defecte enzimatice, este de asemenea important. Cursul deficienței de alfa-N-acetilgalactosaminidază depinde de forma respectivă a bolii.
Boala de Schindler tip I este considerată nefavorabilă, în timp ce prognosticul pentru celelalte două tipuri este mai favorabil. Cu toate acestea, numărul bolnavilor este atât de mic încât nu se pot face declarații semnificative despre speranța de viață.
complicaţiile
Simptomele și complicațiile deficitului de A-N-acetilgalactosaminidază depind foarte mult de forma sa.Cu toate acestea, în cele mai multe cazuri, apar tulburări de mișcare, astfel încât persoanele afectate sunt relativ restrânse în viața de zi cu zi. De asemenea, apar crampe, care sunt asociate cu dureri severe.
Bebelușii suferă în special de tulburări severe de dezvoltare, care pot duce la spasticitate la copil. În unele cazuri, deficiența de A-N-acetilgalactosaminidază duce la orbire completă la pacient. Regresia inteligenței duce la retardare și deci la dizabilități.
Acestea restricționează în mod considerabil viața de zi cu zi a pacientului și au un efect negativ asupra calității vieții. Adesea, persoanele afectate depind de ajutorul altor persoane. Întârzierea poate duce, de asemenea, la o tulburare de găsire de cuvinte sau o tulburare de limbaj. Nu este posibilă tratarea cauzală a deficitului de A-N-acetilgalactosaminidază.
Din acest motiv, persoana afectată trebuie să ia antibiotice. Anumite complicații trebuie de asemenea evitate direct. Pneumonia, de exemplu, este una dintre acestea. Mai mult, pacientul poate depinde și de alimentația artificială.
Când trebuie să te duci la doctor?
Deficitul de A-N-acetilgalactosaminidază trebuie tratat cu siguranță de un medic. Această deficiență nu dispare de la sine și nu există o vindecare spontană a acestei boli. În orice caz, trebuie consultat un medic dacă părinții observă o scădere a abilităților motorii și mentale la copilul sau copilul lor. Reclamațiile cu mișcări sau procese simple pot indica, de asemenea, deficiența de A-N-acetilgalactosaminidază și trebuie examinată de un medic.
Mai mult, diversele spasticități sunt, de asemenea, un semn al unei boli. Nu este neobișnuit ca pacienții să sufere de dizabilități intelectuale și alte defecțiuni. Dacă aceste plângeri sunt prezente, tratamentul este cu siguranță necesar. Dacă nu există tratament, acest lucru poate duce la plângeri și complicații considerabile la vârsta adultă și, astfel, reduce considerabil calitatea vieții persoanei afectate.
Dezvoltarea intensă a vorbirii poate fi, de asemenea, un semn al deficitului de A-N-acetilgalactosaminidază și, prin urmare, trebuie investigată. De regulă, un medic generalist poate fi consultat pentru a determina deficiența de A-N-acetilgalactosaminidază. Tratamentul suplimentar al reclamațiilor individuale este apoi efectuat de un specialist.
Medici și terapeuți din zona dvs.
Tratament și terapie
Nu este posibilă o terapie cauzală pentru deficiența de alfa-N-acetilgalactosaminidază. Prin urmare, abordarea medicală se limitează la tratarea simptomelor. Aceasta include o dietă sensibilă și aportul de lichide, prevenirea bolilor infecțioase, care se poate face și prin administrarea de antibiotice și reținerea atacurilor cerebrale prin administrarea de medicamente anti-epileptice.
Alte etape de tratament includ măsuri de fizioterapie pentru prevenirea pneumoniei și a contracturilor și pentru a reduce durerea sau spasticitatea cu ajutorul medicamentelor. Mai mult, profilaxia prin aspirație poate avea loc prin hrănirea artificială.
Studii recente au arătat că deficiența de a-N-acetilgalactosaminidază este o tulburare de pliere a proteinei, astfel încât terapia de înlocuire a enzimelor este discutată ca o măsură de tratament sensibil. Terapia genică este o altă abordare terapeutică imaginabilă pentru tratamentul tulburărilor de depozitare lizozomală. Deoarece boala Schindler este moștenită într-o manieră recesivă autosomală, se recomandă consilierea genetică a familiilor afectate.
Perspective și prognoză
Deficitul de A-N-acetilgalactosaminidază poate duce la simptome diferite. În majoritatea cazurilor, însă, deficiența duce la dezvoltarea psihomotorie întârziată. La vârsta adultă, pacientul este adesea dependent de ajutorul altor persoane și nu poate face față vieții de zi cu zi de unul singur. De asemenea, apar tulburări de mișcare. Acest lucru poate fi observat printr-un limp sau prin tulburări ale coordonării.
În cel mai rău caz, pacientul poate să orbească sau să sufere de tulburări vizuale severe din cauza deficitului de A-N-acetilgalactosaminidază. Nu este neobișnuit să apară dizabilități intelectuale care reduc considerabil calitatea vieții persoanei în cauză. Apar tulburări de vorbire și de găsire a cuvintelor, care pot afecta viața. Copiii sunt afectați în special de bullying și tachinare din cauza simptomelor deficitului de A-N-acetilgalactosaminidază.
Un tratament cauzal al deficienței de A-N-acetilgalactosaminidază nu este posibil, astfel încât tratamentul are ca scop principal reducerea simptomelor. Se pot utiliza diverse măsuri terapeutice pentru reducerea tulburărilor de dezvoltare. Cu toate acestea, în multe cazuri, pacientul depinde de ajutorul extern din viața de zi cu zi. Speranța de viață nu este afectată de deficiență.
Vă puteți găsi medicamentul aici
➔ Medicamente pentru durereprofilaxie
Nu există măsuri preventive cunoscute împotriva deficitului de alfa-N-acetilgalactosaminidază. Boala Schindler este una dintre bolile extrem de rare.
Dupa ingrijire
Deoarece deficiența de A-N-acetilgalactosaminidază este o boală congenitală care nu poate fi tratată cauzal, ci doar simptomatic, nu este posibil un tratament complet. Persoana afectată depinde de tratamentul pe tot parcursul vieții, motiv pentru care opțiunile pentru îngrijire medicală sunt foarte limitate.
De regulă, datorită deficitului de A-N-acetilgalactosaminidază, pacientul este adesea dependent de aportul de medicamente și antibiotice. Este posibil să se țină seama de posibilele interacțiuni cu alte medicamente, deși antibioticele nu trebuie luate împreună cu alcoolul. Dacă aveți un atac, ar trebui să mergeți la spital sau să apelați direct la un medic de urgență.
Mai mult decât atât, plămânii trebuie economisiți pentru a evita inflamația. Prin urmare, pacienții cu deficiență de A-N-acetilgalactosaminidază nu ar trebui să fumeze în niciun caz. O dietă sănătoasă și un stil de viață în general sănătos au un efect foarte pozitiv asupra evoluției bolii.
Dacă pacientul dorește să aibă copii, consilierea genetică este utilă pentru a preveni transmiterea bolii la copii. Deoarece pacienții suferă adesea de mobilitate restrânsă, fizioterapia este utilă pentru a atenua acest lucru. Multe exerciții pot fi, de asemenea, efectuate în propria casă.
Puteți face asta singur
O deficiență de A-N-acetilgalactosaminidază existentă este o boală incurabilă. Prin urmare, tratamentul medical este esențial. Măsurile de auto-tratament se pot baza doar pe simptome pentru a facilita viața de zi cu zi. Este important ca părinții să observe copilul îndeaproape.
Părinții își pot ajuta cel mai bine copilul prin înțelegere și răbdare, dragoste și grijă. Mai mult, tratamentele de terapie ocupațională și logopedie se bazează întotdeauna pe un concept dublu: terapia la fața locului cu specialistul și efectuarea exercițiilor la domiciliu. Părinții ar trebui să fie activi aici în mod consecvent și cu răbdare. Trebuie asigurată o dietă bogată în vitamine și minerale, exerciții fizice regulate și mult aer proaspăt. Acest lucru întărește sistemul imunitar și reduce riscul de infecție. În cazul în care persoana are tendința de a se confunda, nu trebuie să fie singură mult timp și trebuie verificată pentru a vedea dacă se poate răni singură dacă are loc o convulsie.
De cele mai multe ori, pacienții nu pot face față vieții de zi cu zi și necesită îngrijiri constante. Dacă părinții nu își permit acest lucru - în special cu copiii mai mari și adulții - nu ar trebui să le fie frică să caute ajutor. Aceasta poate lua forma unei asistente medicale sau a unui plasament într-o unitate adecvată. Dacă mai doriți să aveți copii, este recomandat un consult cu un specialist, deoarece boala este moștenită.
.jpg)










.jpg)