Boala metabolică ereditară Fenilcetonuria (PKU) apare rar, dar în cazul unei boli a copilului, necesită o nutriție constantă încă din primul minut pentru a preveni deteriorarea dezvoltării creierului și a complicațiilor care apar cu acesta.
Ce este fenilcetonuria?
.jpg)
© bacsica - stock.adobe.com
Fenilcetonuria este o boală metabolică ereditară în care se acumulează o anumită componentă proteică în organism, care restricționează în primul rând dezvoltarea creierului la copii.
În Republica Federală Germania, un singur copil din 10.000 este afectat statistic. Dacă acest copil este diagnosticat cu boala precoce, fenilcetonuria poate fi tratată și copilul se poate dezvolta complet normal.
cauze
Cauza pentru care Fenilcetonuria este un defect genetic. Aceasta înseamnă că componenta proteică fenilalanină, care apare în toate alimentele care conțin proteine și, prin urmare, este ingerată cu alimente, nu mai poate fi defalcată. La persoanele sănătoase, o enzimă funcționează în acest scop, care la persoanele cu fenilcetonurie, în funcție de gravitate, funcționează doar parțial sau a încetat să funcționeze complet.
Simptome, afectiuni si semne
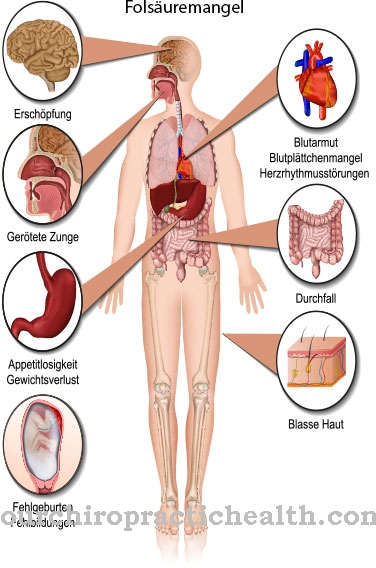
Dacă afecțiunea nu este diagnosticată și tratată imediat după naștere, primele semne vor apărea în jurul vârstei de trei luni. Maturizarea creierului este perturbată, creierul nu crește adecvat, ceea ce este vizibil la copiii mai mari de către un cap semnificativ mai mic. Dezvoltarea mentală rămâne în urmă. Când ajung la pubertate, au handicapuri intelectuale severe.
Datorită afectării celulelor creierului, copiii suferă de hiperexcitabilitate, ceea ce cauzează adesea convulsii epileptice. Celulele musculare sunt de asemenea afectate de boală. Mușchii se strâng ca spasmele (spasticitatea), ceea ce îngreunează mișcarea normală. Mai mult, apar tulburări de comportament sub formă de hiperactivitate și agresivitate. Copiii sunt predispuși la izbucniri necontrolate de furie.
Un semn tipic de PKU este mirosul de acetonă emis de cei afectați. Este cauzată de formarea acetatului de fenil în organism. Această substanță este excretată prin glandele sudoripare și este, de asemenea, conținută în urină. Aspectul celor care suferă de PKU este caracteristic.
Deoarece oranismul lor nu poate produce suficient melanina pigmentară, ele au adesea părul blond alb, pielea foarte ușoară, sensibilă și ochii albastru deschis, strălucitori. Tulburarea de pigmentare poate provoca erupții cutanate asemănătoare eczemelor. Gravitatea simptomelor depinde de cât de sever este metabolizat și se abate de la normă.
Diagnostic și curs
Fenilcetonuria poate fi determinat printr-un test de sânge, care este de obicei efectuat la screeningul nou-născutului ca parte a așa-numitei examinări preventive U2. Testul de sânge efectuat aici va determina, printre altele, nivelul de fenilalanină al sugarului.
Deoarece metabolismul este perturbat într-o boală cu fenilcetonurie și fenilalanina nu mai poate fi descompusă, este depus din ce în ce mai mult în sânge și poate fi detectat aici. O valoare crescută de fenilalanină de 1 până la 2 miligrame per decilitru în sânge indică o boală cu fenilcetonurie.
Dacă se suspectează fenilcetonurie în timpul sarcinii, un test de lichid amniotic poate fi utilizat pentru a efectua un test ADN prenatal la sugar și pentru a face un diagnostic. Dacă fenilcetonuria este diagnosticată, se vor face teste suplimentare pentru a determina severitatea afecțiunii și se va viza tratamentul.
Dacă fenilcetonuria nu este tratată, primele simptome ale bolii metabolice apar după aproximativ trei luni, care duc rapid la complicații grave, inclusiv dezvoltarea psihică întârziată la dizabilități intelectuale severe, tulburarea musculară și chiar convulsii epileptice.
complicaţiile
În orice caz, fenilcetonuria trebuie tratată imediat după nașterea copilului. Dacă acest tratament nu este efectuat, poate duce la plângeri grave în timpul creșterii și dezvoltării copilului, care nu pot fi compensate ulterior. Cei afectați, de obicei, suferă de o tulburare metabolică din cauza fenilcetonurie.
De asemenea, duce la o iritabilitate crescută, astfel încât copiii să pară agresivi. Întârzierea dezvoltării motorii și mentale poate deveni, de asemenea, evidentă și poate reduce semnificativ calitatea vieții persoanei în cauză. În multe cazuri, părinții suferă și de depresie sau alte tulburări psihologice. Cei afectati prezinta, de asemenea, un miros neplacut al corpului, astfel incat copiii si adolescentii afectati pot deveni victime ale tachinarii sau intimidarii.
Mai mult, apar convulsii epileptice, care în cel mai rău caz pot duce la moarte. Tulburările de pigment pot apărea și la copil. Tratamentul pentru fenilcetonurie nu este asociat cu complicații. Alimentația corectă poate preveni evoluțiile nedorite, astfel încât să nu mai existe complicații la vârsta adultă.
Când trebuie să te duci la doctor?
Deoarece fenilcetonuria poate duce la deteriorarea severă a creierului copilului, această boală trebuie tratată imediat în orice caz. Un diagnostic precoce este foarte important pentru a preveni complicațiile ulterioare sau dezvoltarea afectată a copilului. De regulă, fenilcetonuria se manifestă ca o dezvoltare mai lentă a copilului, dizabilitățile intelectuale și convulsiile epileptice joacă și ele.
În cazul unei crize epileptice, apelați un medic de urgență sau mergeți imediat la spital, deoarece aceasta este singura modalitate de a evita daunele suplimentare. La o vârstă fragedă, fenilcetonuria duce adesea la izbucniri de furie sau tulburări de comportament severe. Dacă apar și aceste simptome, este necesară o vizită la medic. Tulburările sau petele pigmentare de pe piele pot indica și această boală.
Tratamentul fenilcetonurie depinde de simptomele exacte și este efectuat de diverși specialiști. Tratamentul psihologic, la care pot participa și părinții sau rudele, este adesea util. În cazul diagnosticului și tratamentului precoce, speranța de viață a pacientului nu este de obicei afectată negativ.
Tratament și terapie
Când un Fenilcetonuria a fost diagnosticat, de obicei poate fi tratat foarte bine, astfel încât copiii să nu fie nevoiți să sufere de o dezvoltare anormală a creierului și, în schimb, să se dezvolte complet normal mental.
Cu toate acestea, acest lucru este posibil numai cu o dietă respectată în mod constant la dieta scăzută de fenilalanină. Nu există nicio altă opțiune terapeutică, de exemplu pe bază de medicamente, pentru a trata fenilcetonuria și pentru a evita deteriorarea creierului. Este deosebit de important să se evite aproape complet alimentele care conțin fenilalanină în perioada în care creierul se dezvoltă - adică în perioada de la naștere până la pubertate.
Cu cât dieta începe mai devreme, cu atât progresia fenilcetonurie este mai bună. Din acest motiv, dieta este de obicei începută imediat ce se naște un nou-născut, iar în locul laptelui matern, sugarii li se oferă o formulă specială care conține cu greu fenilalanină. Chiar dacă o dietă săracă în fenilalanină este deosebit de importantă până la pubertate, medicii recomandă o dietă pe tot parcursul vieții cu conținut scăzut de fenilalanină pentru a nu fi restricționată de boală la bătrânețe.
Cu toate acestea, în dietă este important pentru tratamentul fenilcetonurie ca dieta să conțină puțin fenilalanină, dar să nu împiedice complet absorbția fenilalaninei. Deoarece fenilalanina este un aminoacid vital care este deosebit de necesar pentru creșterea copiilor care suferă de fenilcetonurie. Cu toate acestea, nu trebuie depășită o anumită valoare în sânge. Prin urmare, persoanele cu fenilcetonurie trebuie să facă teste de sânge regulate.
Perspective și prognoză
Prognosticul pentru fenilcetonuria clasică este extrem de bun dacă diagnosticul este făcut din timp, dacă este posibil la nou-născutul. Dacă o dietă cu fenilalanină scăzută este administrată la începutul copilului, nu trebuie să se aștepte deficiențe fizice sau psihice. Cei afectați au o viață normală și o speranță medie de viață.
O condiție importantă pentru un prognostic pozitiv este respectarea strictă a dietei, în special în primii șase ani de viață, când creierul se dezvoltă deosebit de bine. Femeile cu fenilcetonurie pot de asemenea să rămână însărcinate și să aibă copii proprii. Deși este o boală metabolică ereditară, nu este de așteptat niciun rău pentru copil. Cu toate acestea, dieta trebuie urmată constant în timpul sarcinii.
Fără dieta scăzută de fenilalanină, boala duce la tulburări ale dezvoltării creierului în copilărie timpurie. Anomaliile neurologice pot fi deja recunoscute începând cu a patra lună de viață. Deoarece afectarea creierului este ireversibilă, nu există nicio perspectivă de vindecare. Coeficientul de inteligență va fi, de regulă, permanent sub norma. În plus, pot apărea convulsii, tulburări de comportament și dizabilități motorii.
Un caz special este forma atipică de fenilcetonurie, care se caracterizează printr-o deficiență în coenzima BH4 (tetrahidrobiopterină). Prognosticul dezvoltării intelectuale la acești pacienți nu poate fi evaluat în mod constant. În ciuda unei diete timpurii, pot apărea leziuni ale sistemului nervos.
profilaxie
Întrucât este un Fenilcetonuria Dacă este o boală metabolică ereditară, nu există nici o modalitate de a preveni fenilcetonuria. Cu toate acestea, dacă aveți fenilcetonurie, o dietă cu alimente sărace în fenilalanină poate preveni simptomele și complicațiile care apar de obicei cu fenilcetonuria.
Pentru a-și proteja proprii copii de consecințele unei boli cu fenilcetonurie, persoanele care sunt deja însărcinate, al căror partener este însărcinat sau care intenționează să aibă o sarcină ar trebui să respecte cu strictețe o dietă cu fenilalanină scăzută. Dacă copilul se îmbolnăvește, trebuie să se acorde atenție unei diete sărace în fenilalanină pentru bunăstarea copilului.
Dupa ingrijire
Întrucât fenil-kentonuria este una dintre afecțiunile metabolice ereditare și nu poate fi vindecată, ci doar tratată, îngrijirea ulterioară și tratamentul sunt în mare măsură identice. Aceasta include în primul rând o dietă scăzută de fenilalanină și monitorizarea periodică a nivelului de PA în sânge. Aceste măsuri trebuie respectate strict până la vârsta de șase ani, dar sunt necesare pentru viață.
Cu fenilcetonuria atipică, coenzima BH4 trebuie de asemenea completată. Dacă tratamentul a fost început prea târziu, îngrijirea de urmărire se poate extinde și la daunele existente. Acestea sunt în mare parte în zona de dezvoltare a creierului și pot varia ca gravitate.
Începerea timpurie a măsurilor de sprijin (de exemplu, terapia ocupațională) poate fi utilă pentru a combate tulburările de comportament. Acestea includ probleme de somn, nevoia de control, comportamentul auto-dăunător sau antisocial, a cărui gravitate poate fi adesea atenuată cu succes. În cazul tulburărilor motorii, fizioterapia vizată poate îmbunătăți simptomele.
Dacă se respectă măsurile prescrise, acum este posibil ca mulți pacienți să ducă o viață în mare parte normală. Când ajungi la pubertate, cea mai sensibilă fază s-a terminat. De obicei, nu mai sunt de așteptat defecte neurologice mai mari, chiar dacă fluctuațiile nivelului PA pot afecta temporar chimia creierului. Aportul de preparate de dopamină ajută împotriva acestui lucru.
Puteți face asta singur
Cu fenilcetonuria clasică, aderarea la o dietă vegană pe tot parcursul vieții, fără a fi asociată sursa de fenilalanină, este în prim plan. Nou-născuții sunt hrăniți exclusiv cu formulă fără fenilalanină. Paharele de fructe și legume sunt potrivite ca alimente complementare. Părinții copiilor care suferă de PKU trebuie să calculeze conținutul de fenilalanină din toate produsele alimentare pe care le hrănesc și să respecte în mod constant valorile limită.
Ulterior, mâncarea pentru bebeluși va fi înlocuită cu o pulbere proteică special dezvoltată pentru această boală. La fiecare masă trebuie adăugat amestecul de aminoacizi pentru pacienții cu PKU. Formează componenta principală de-a lungul vieții a dietei necesare cu un nivel scăzut de fenilalanină. Prin urmare, este recomandabil să-l obișnuim pe copilul cu mirosul și gustul pulberii într-un mod jucăuș.
Există acum o gamă largă de cursuri de instruire și cursuri de gătit pentru copiii afectați de toate vârstele. Aceste măsuri pedagogice promovează treptat responsabilitatea personală. În același timp, susțin o manipulare pozitivă a restricțiilor nutriționale. Produsele pregătite cu un conținut scăzut de fenilalanină permit pacienților tineri să participe la excursii și excursii școlare. Părinții copiilor bolnavi sunt expuși la stres organizațional și psihologic sever. Este util pentru ei să facă schimb de idei cu ceilalți suferinzi în grupuri de ajutor.
Pacienții adulți cu fenilcetonurie care doresc să aibă copii trebuie să aibă grijă deosebită atunci când își planifică familiile: doar respectarea unei diete stricte asigură nivele normale de fenilalanină în timpul sarcinii. Valorile normale sunt absolut necesare în momentul concepției pentru a evita deteriorarea gravă a copilului nenăscut.



.jpg)
.jpg)
.jpg)




.jpg)