Hipoxantina este o purină deviată și are formă legată ca nucleobază și în formă liberă, de ex. B. în urină. Se găsește și în glande și măduvă osoasă. Ca produs de dezaminare a adeninei, hipoxantina este oxidată la acid uric și xantină. Mai rar formează o structură de bază a acizilor nucleici.
Ce este Hypoxanthine Guanine Fosforibosiltransferaza?
Enzima tetramerică este formată din hipoxantină și guanină Hypoxanthine guanine fosforibosil transferaza.
Tetramerele sunt macromolecule care constau din patru blocuri de construcții similare, mai precis din monomeri. Enzima este una dintre cele mai importante în metabolismul purin al eucariotelor, este sensibilă la schimbările genice și poate provoca devieri la om prin mutații genice, care sunt exprimate în anumite boli metabolice. Astfel sunt z. B. Sindroamele Lesch-Nyhan și Kelley-Seegmiller.
Funcție, efect și sarcini
Enzima hipoxantină-guanină-fosforibosiltransferază crește metabolismul purinei și eficacitatea sa energetică.
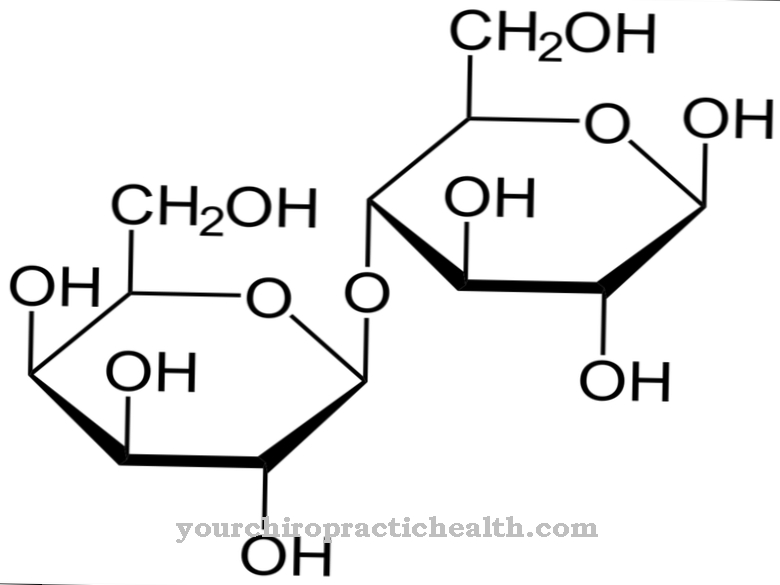
Aceasta se bazează pe baze de purină, care sunt acizi nucleici care sunt structural derivați de purină. Este vorba despre xantina, hipoxantina, adenina și guanina, care se atașează de alte baze prin legături de hidrogen. Astfel de legături au o influență majoră asupra dublei helixuri și replicării ADN-ului și joacă un rol în biosinteza proteinelor.
Bazele purine pot fi reciclate de două enzime. În plus față de hipoxantină guanină fosforibosil transferază, aceasta este adenina fosforibosil transferază. Ambele formează un nucleotid printr-un reziduu de fosforibosil, care la rândul său este un bloc de bază de acizi nucleici atât în ADN cât și în ARN. Molecula este formată din zahăr, bază și fosfat și controlează funcțiile de reglare vitale în celule. În timpul acumulării, ATP este economisit și formarea acidului uric este redusă.
Dacă bazele purinice sunt reciclate, este denumită calea de salvare. Acesta este un termen general pentru căile metabolice în care o sinteză a biomoleculelor rezultă din produsele de degradare. Organismul își desfășoară propriul proces de reciclare, prin care aproximativ nouăzeci la sută din bazele purine sunt refolosite și zece la sută sunt de fapt excretate. Acest lucru arată eficiența reciclării bazei purine și importanța hipoxantinei-guaninei fosforibosil transferază.
Educație, apariție, proprietăți și valori optime
Dacă apar mutații în gena HPRT, dimensiunea și aminoacizii se pot schimba. Aceasta poate fi încorporarea unor secvențe ADN suplimentare sau nucleotide, ceea ce la rândul său duce la o producție incorectă a produsului genic care este codat pe gena respectivă, sau chiar la ștergerea întregii secvențe. Este z. B. schimbă secvența de aminoacizi, apar boli precum guta.
Bolile metabolice, cum ar fi sindromul Lesch-Nyhan, ca urmare a unui defect genetic, sunt deosebit de grave. Aceasta este moștenită într-o manieră recesivă legată de x, ceea ce înseamnă că afectează mai ales bărbații care au un singur cromozom X. Defectul genetic poate fi prezent la femei, dar se declanșează doar ca boală atunci când ambii cromozomi X sunt afectați, ceea ce este relativ rar. Cel mai adesea al doilea cromozom X compensează defectul primului.
Vă puteți găsi medicamentul aici
➔ Medicamente pentru sănătatea vezicii urinare și a tractului urinarBoli și tulburări
Sindromul se manifestă printr-o deficiență de hipoxantină guanină fosforibosil transferază. Enzima nu este produsă din cauza defectului genetic. Datorită mutației și lipsei de reciclare și transformare a bazelor de guanină și hipoxantină, există o acumulare de baze purine care trebuie să fie construite și excretate de organism.
Defalcarea are loc prin xantina produsului intermediar, care este transformat în acid uric și excretat prin rinichi. Dacă acest proces este restricționat, în zona articulațiilor se formează cristale de acid uric, care declanșează mai multe atacuri de gută. Enzima nu mai este produsă, nivelul acidului uric din țesut și sânge crește, iar sistemul nervos central este deranjat.
Sindromul Lesch-Nyhan nu este vizibil direct la naștere. O poziție vizibilă a picioarelor și tendința copilului de a se mișca puțin și de a se dezvolta mai lent se poate observa numai după aproximativ zece luni. Sindromul este slab și sever. O secreție crescută de acid uric și atacuri de gută mai ușoare sunt forma mai ușoară, cu simptome severe, aceasta vine la auto-vătămare, deficiență mentală severă și agresivitate. Auto-rănirea apare prin mușcăturile degetelor sau ale buzelor. Când mușcăm în extremități, se poate observa adesea că persoanele afectate își limitează autoagresiunea la o singură mână. La rândul său, agresiunea este adesea îndreptată împotriva persoanelor apropiate, cum ar fi frații sau părinții.
Cea mai gravă formă a bolii se caracterizează printr-o disfuncție neurologică multiplă și o tendință foarte accentuată spre auto-mutilare. Sindromul se manifestă prin spasticitate, distonie, hipotonie, coreoreatoză și o disponibilitate crescută de reacție la reflexe. Calitățile și dezvoltarea mentală sunt sever restricționate. În această afecțiune, sindromul poate duce și la moarte într-o măsură deosebit de drastică.
Boala este diagnosticată printr-un tablou medical. Nivelul acidului uric din urină și sânge este măsurat și activitatea hipoxantinei-guaninei fosforibosiltransferază în țesut și sânge. Acesta din urmă este mult redus și poate fi prezent și prenatal.
Tratamentul bolii este dificil. Cura nu este posibilă și fără tratament copilul moare în primii ani de viață. În unele cazuri, dinții bebelușului trebuie extrasi ca măsură preventivă. Alte abordări terapeutice includ scăderea nivelului acidului uric folosind medicamente precum alopurinolul, care acționează ca un inhibitor al gutei. Bazele purine nu sunt reciclate, dar acidul uric este defalcat mai bine. Tulburările respective, infecțiile și leziunile nervoase sunt, de asemenea, tratate și se recomandă o dietă specială, care de obicei nu conține carne și are un nivel scăzut de purină.
De asemenea, se efectuează cercetări cu privire la efectele secundare psihosomatice ale stimulării profunde a creierului. Medicina speră că acest lucru va preveni agresiunea și auto-mutilarea. Sindromul Kelley-Seegmiller, pe de altă parte, este cea mai ușoară formă a unei deficiențe în hipoxantină-guanină fosforibosil transferază. Tot aici se produce prea mult acid uric și apar boli de gută precoce. Primele indicii ale sindromului sunt cristale de portocale în scutecul copilului, infecții ale tractului urinar și urolitiaza. Guta sau artrita acută se dezvoltă în perioada pubertății.
O subdezvoltare mentală și auto-atacuri, așa cum apar cu sindromul Lesch-Nyhan, nu sunt cazul, cel mult poate duce la tulburări de atenție. Tratamentul precoce le permite, de obicei, persoanelor afectate să aibă o speranță de viață normală.








.jpg)