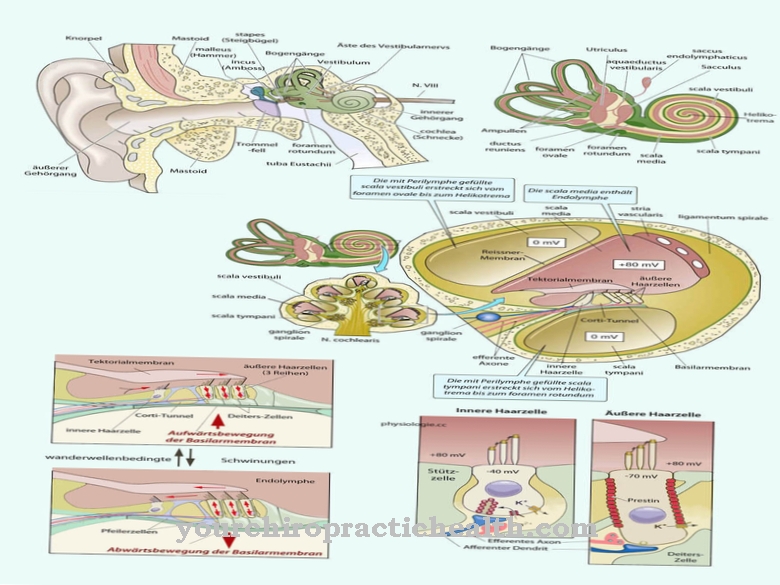
În total, sunt 45 diferite boli de depozitare lizozomale, care sunt un grup eterogen de boli metabolice congenitale. Persoanele care suferă de oricare dintre aceste boli au un defect genetic. Toate bolile de depozitare au un lucru în comun: o anumită enzimă este fie absentă, fie doar parțial funcțională.
Ce este boala de depozitare lizozomală?

© designua - stock.adobe.com
Aceste boli congenitale de depozitare sunt rare, deoarece mai puțin de cinci din 10.000 de persoane sunt afectate. Diferitele boli au un curs foarte diferit, iar simptomele pot varia foarte mult.
Cele mai cunoscute forme de boala de depozitare lizozomală sunt boala Fabry, boala Gaucher, boala Pompe și mucopolizaharidoza (MPS). Ele sunt adesea denumite „orfani ai medicamentului”, deoarece calea către un diagnostic specific și o terapie adecvată poate fi foarte lungă. Uneori, poate fi nevoie de ani de zile pentru ca cei afectați să afle ce li se întâmplă.
cauze
Bolile de depozitare lizozomiale se caracterizează prin anumite forme de boli metabolice ereditare. Pacientului îi lipsește o enzimă importantă care asigură buna echilibrare metabolică. În forma mai puțin pronunțată, această enzimă nu este cel puțin prezentă în cantități suficiente.
Sarcina enzimelor este de a elimina substanțele poluante și deșeurile care se acumulează în organismul uman prin intermediul metabolismului prin lizozomi sau de a le procesa din nou, astfel încât să nu apară simptome.
Dacă există o deficiență de enzimă, acest ciclu de eliminare funcțională fără probleme nu mai este garantat. Substanțele dăunătoare se instalează în celule și perturbă ciclul metabolic. În faza inițială, tulburările nu au un efect vizibil, există doar câteva restricții. Cu toate acestea, dacă această tulburare metabolică rămâne netratată ca urmare a unei deficiențe enzimatice, simptomele se înmulțesc, deoarece celulele devin foarte mari.
Simptome, afectiuni si semne
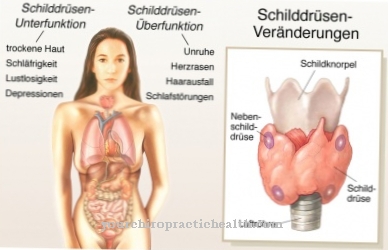
În cel mai rău caz, acestea se aplică. Consecințele sunt deteriorarea oaselor, sistemului nervos, splinei, rinichilor, mușchilor sau inimii. Datorită activității enzimei reduse sau absente, boala Fabry determină depozitarea de grăsimi (globotriaozilceramidă, Gb3) în celule. Aceste depuneri nedorite pot duce la dureri severe la nivelul degetelor de la picioare sau deget, accident vascular cerebral și leziuni renale.
Diagnosticul și cursul bolii
Această imagine clinică afectează diferite sisteme în același timp: vasele de sânge, rinichii, inima și sistemul nervos. Boala Gaucher, moștenită autosomală recesivă, provoacă o mutație a enzimei "beta-glucocerebrosidaza" și duce la o acumulare de substrat în celule, în special în macrofage (fagocite) care aparțin sistemului reticulo-endotelial. Numărul de sânge se modifică, ficatul și splina se măresc, iar oasele doare.
Boala este progresivă și este în mare parte etnică, deoarece apare în majoritatea cazurilor la persoane de origine evreiască. Boala Pompe este cunoscută și sub denumirea de „deficit de acid maltază”. Tabloul clinic aparține grupului de glicogeneză de tip II. Persoanele afectate nu au enzima "alfa-1,4-glucozidaza" (acid maltaza) sau nu sunt disponibile în cantități suficiente. Datorită degradării glicogenului afectat în mușchi, pacienții suferă de distrugerea celulelor musculare sub formă de depozitare a zahărului.
Mucopolizaharidoza tip I (MPS), cunoscută și sub numele de boala Hunter, are diverse cauze clinice. Boala Hurler este cea mai severă formă, iar boala Scheie se află la sfârșitul patogenezei clinice. Există tranziții cu caracteristici diferite între aceste două forme de progresie. Cea mai proeminentă caracteristică este defalcarea descompunerii carbohidraților care se acumulează în lizozomii celulelor.
Pacienții cu boală vânătorilor pot prezenta statură scurtă, splină și ficat lărgite, trăsături grosiere, piele îngroșată, limbă mărită și dificultăți de respirație. În plus, scheletul este adesea schimbat în zona pelvisului, a coloanei vertebrale, a oaselor mâinilor și a craniului. Sunt posibile ombilicale și [[hernii inghinale].
complicaţiile
În majoritatea cazurilor, simptomele sau complicațiile apar foarte târziu în această boală. Din această cauză, este diagnosticat cu întârziere, astfel încât tratamentul precoce nu este posibil în majoritatea cazurilor. Fără tratament, diverse plângeri și leziuni ale organelor interne apar pe măsură ce boala progresează.
Rinichii, ficatul și splina sunt afectate în special. Inima poate fi afectată și de această boală, care poate duce la moarte cardiacă în cel mai rău caz. Mai mult, se produc leziuni la rinichi, iar cei afectați suferă adesea de durere în degetele de la picioare sau deget. Paraliza poate apărea și dacă creierul a fost afectat de această boală. Ficatul și splina pot fi măriți și pot provoca dureri severe.
Nu este neobișnuit ca oasele persoanei afectate să fie fragile și, de asemenea, dureroase. Tratamentul acestei boli se dovedește dificil. În multe cazuri, speranța de viață a persoanei afectate este redusă semnificativ. De obicei, nu există complicații particulare atunci când utilizați medicamente. Cu toate acestea, un curs pozitiv al bolii nu poate fi garantat în niciun caz.
Vă puteți găsi medicamentul aici
➔ Medicamente pentru durereCând trebuie să te duci la doctor?
Căderea părului, problemele articulare și tulburările de organ sunt posibile semne ale bolii de depozitare lizozomală. Se recomandă o vizită la medic dacă simptomele continuă să reapară sau apar brusc, fără a fi găsită o cauză. Dacă simptomele sunt legate de un defect enzimatic deja diagnosticat sau de o altă boală gravă, trebuie consultat medicul responsabil. O boală de tratare netratată poate duce la demență, infertilitate, neuropatii și alte complicații, unele dintre ele punând în pericol viața. Prin urmare, toate simptomele imaginabile ar trebui examinate, chiar dacă nu există suspiciuni specifice.
Simptomele bolii de depozitare lizozomală pot apărea în faze sau se pot dezvolta insidios, dar necesită întotdeauna examinare și tratament. Persoanele afectate sunt cel mai bine să vorbească direct cu medicul de familie sau cu un internist. Terapia propriu-zisă are loc de obicei într-o clinică de specialitate pentru boli interne, deși fizioterapia sau psihoterapia pot fi conectate în funcție de simptome. În special, măsurile terapeutice sunt indicate datorită cursului adesea negativ al bolii.
Terapie și tratament
În funcție de cât de devreme se pune un diagnostic adecvat, aceste boli ereditare pot fi tratate foarte bine cu terapia de înlocuire a enzimelor, astfel încât persoanele afectate să aibă mult mai puține simptome și, astfel, o calitate mai bună a vieții. Această terapie de înlocuire este utilizată în conformitate cu tabloul clinic.
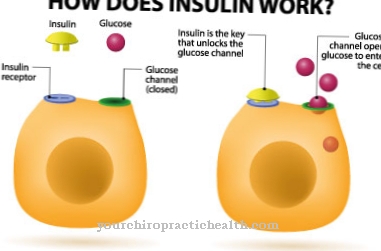
Persoanelor care suferă de boala Gaucher le lipsește „enzima ß-glucocerebrosidaza”, care este produsă biotehnologic și infuzată în corpul pacientului. Lisozomii acționează eficient și sunt capabili să absoarbă substanțele din mediul lor imediat. Din acest motiv, enzimele utilizate artificial sunt modificate astfel încât pot fi furnizate lizozomilor într-un mod ideal.
Macrofagele (fagocitele) descompun glucocerebrosidele care s-au acumulat în celule. Această terapie poate fi comparată cu insulinoterapia pentru diabetul zaharat, cu diferența că nu este un hormon lipsă, ci o enzimă inexistentă care este furnizată. Organismul descompune în mod regulat toate substanțele, inclusiv enzima artificială furnizată.
Din cauza acestei descompuneri periodice a substanței, pacienții trebuie să urmeze această terapie perfuzabilă în mod regulat până la sfârșitul vieții. Terapia de înlocuire a enzimelor nu acționează simptomatic, ci combate direct cauza bolii ereditare. Medicii numesc această terapie cauzală. Principiile terapiei trebuie utilizate pentru toate cele patru boli de depozitare comune menționate anterior.
De asemenea, pacienții cu pompe sunt tratați cu perfuzie. În această boală, enzima inexistentă "acid alfa glucosidaza" este furnizată și ajută la descompunerea glicogenului care s-a acumulat în lizozomii mușchilor. La pacienții cu tipul de boală „mucopolizaharidoză tip I”, enzima lizozomală „alfa-iduronidaza” nu este prezentă sau nu este prezentă în cantități suficiente. Este una dintre cele mai rare boli de stocare în care se acumulează molecule de zahăr în organe și țesuturi.
Dacă procesul este normal, enzima descompune mucopolizaharidele. Moleculele de zahăr sunt cu lanț lung și sunt implicate în dezvoltarea țesutului de susținere și conectiv, de exemplu oase, piele, lichide articulare și cartilaj. Dacă cursul normal al degradării este perturbat din cauza lipsei enzimei, glicozaminoglicani patologici (GAG) se acumulează în celulele individuale. Opțiunile de terapie viitoare au ca scop administrarea de tablete.
Perspective și prognoză
Prognosticul pentru boala de depozitare este slab. S-a constatat că o dispoziție genetică este cauza bolii de sănătate. Cerințele legale interzic medicii și oamenii de știință să schimbe genetica umană. Din acest motiv, boala rămâne toată viața și nu are nicio perspectivă de recuperare.
Medicul curant se concentrează pe tratarea simptomelor care apar. Dacă sunt lăsate netratate, diverse reclamații vor crește în timp. Sistemul osos este deteriorat și apar probleme ale organelor. În cel mai rău caz, organele interne vor funcționa defectuos și, în final, funcția lor va eșua. Aceasta amenință persoana în cauză cu moartea prematură.
Provocarea bolii constă în diagnosticare. La un număr mare de pacienți, reclamațiile notabile și puternic perceptibile apar doar mai târziu în viață. Drept urmare, tulburarea genetică rămâne neobservată mult timp, iar tratamentul precoce al bolii este dificil. Cu cât se pune mai târziu un diagnostic, cu atât este mai nefavorabil cursul ulterior. Într-un stadiu avansat al bolii, organele sau articulațiile interne sunt deja grav afectate. Sunt necesare intervenții chirurgicale și dacă boala progresează nefavorabil, doar un organ donator poate salva viața persoanei afectate. Prin urmare, tratamentul precoce este esențial pentru un prognostic îmbunătățit.
profilaxie
Deoarece este un defect genetic congenital care împiedică exprimarea unei enzime, această boală nu poate fi tratată preventiv. Cu toate acestea, cele mai recente realizări ale ingineriei genetice ar putea oferi o abordare în acest domeniu.
Dupa ingrijire
Cu această boală, oamenii suferă de o serie de complicații și afecțiuni diferite. De regulă, toate acestea au un efect foarte negativ asupra calității vieții persoanei afectate, astfel încât un diagnostic trebuie făcut foarte devreme. Cu cât este consultat mai devreme un medic, cu atât este mai bine cursul ulterior al acestei boli.
Severitatea acestei boli poate varia foarte mult, astfel încât adesea nu este posibilă o predicție generală. Cei afectați suferă de leziuni severe organelor interne. Rinichii și inima sunt afectate în primul rând, astfel încât copilul poate muri în primele zile dacă simptomele nu sunt corectate la timp. Există, de asemenea, depozite de grăsimi în diferite părți ale corpului.
Degetele și degetele de la picioare sunt afectate în special, ceea ce poate duce la o estetică semnificativ redusă pentru persoana afectată. De regulă, deteriorarea rinichilor și a creierului apare în cursul următor, astfel încât persoana afectată moare ca urmare a acestei afectări. Părinții și rudele suferă adesea de depresie sau alte tulburări mintale din cauza bolii.
Puteți face asta singur
Bolile de depozitare lizozomică necesită foarte des îngrijiri medicale intense. Adesea nu există suficiente oportunități de autoajutorare. Părinții copiilor afectați de multe ori se confruntă cu stres sever în mediul de origine, deoarece copilul lor are nevoie de îngrijire și atenție constantă.
Imaginile clinice ale bolilor individuale de depozitare sunt diferite. Există atât forme ușoare, cât și foarte dificile. Un exemplu este boala lui Gaucher. Ajutorul părinților este adesea limitat la hrănirea copilului cu handicap sever. În cazuri mai blânde, speranța de viață poate fi aproape normală. Cu toate acestea, supravegherea medicală constantă este necesară pentru a evita posibilele complicații. Activitatea fizică regulată este una dintre terapiile de însoțire care pot fi efectuate și acasă. În plus, trebuie să fie organizat un examen amănunțit de screening pentru cancer. Aceasta necesită vizite continue la medic cu copilul lor de la părinți. Același lucru este valabil și pentru alte boli de depozitare lizozomale.
În cazul unor boli, pe lângă dizabilitățile fizice, pot apărea și deficiențe mintale, care necesită în continuare un sprijin special. În forme mai blânde ale anumitor boli, cum ar fi boala Hunter, inițial apar doar modificări ale scheletului și dismorfisme faciale. Totuși, aici, pacientul afectat este adesea capabil să ducă o viață independentă. Cu toate acestea, aici sunt necesare și examene medicale constante pentru a exclude posibile complicații, cum ar fi insuficiența cardiacă sau boli respiratorii. Pacientul poate face față stresului psihologic cauzat de deformații fizice prin consiliere psihologică.
.jpg)
.jpg)




.jpg)





.jpg)