Sindromul Bardet-Biedl, de asemenea Sindromul Laurence-Moon-Biedl-Bardet (LMBBS), este o boală din domeniul ciliopatiilor care apare exclusiv datorită eredității. Sindromul se manifestă sub formă de malformații multiple care sunt declanșate de modificări (mutații) pe diferite locații ale genelor sau cromozomi.
Ce este sindromul Bardet-Biedl?

© Imagini Creativa - stock.adobe.com
Tabloul clinic definit de medicii Moon și Laurence și mai târziu de Bardet și Biedl este o boală în care distrofia retiniană apare ca o caracteristică semnificativă din punct de vedere medical în combinație cu alte simptome. Datorită acestei situații medicale inițiale complicate, determinarea finală a bolii BBS este dificilă. Această imagine clinică a fost înregistrată medical pentru prima dată în 1866.
Patru persoane examinate au avut retinită pigmentă (distrofie retinală, RP) în asociere cu paraplegia (paralizie spastică), precum și hipogenitalism (organe genitale subdezvoltate) și un handicap mental. În 1920, medicul francez Bardet a descris o boală care a fost compusă din RP (distrofie retinală), hipogenitalism, polidactilie și obezitate.
Patologul din Praga, Biedl, a descoperit și debilitate (confuzie mentală). În 1925, cercetătorii Weiss și Solis-Cohen au rezumat cazurile cunoscute și au descris tabloul clinic ca fiind Sindromul Laurence-Moon-Biedl-Bardet.
cauze
În anii care au urmat, literatura medicală a subliniat din ce în ce mai mult că cazurile înregistrate de Laurence și Moon sunt o formă specială rară care apare doar în cazuri izolate împreună cu BBS. Rezultatele cercetărilor medicale mai recente atribuie sindromul Bardet-Biedl zonei ciliopatiilor (boli ciliare).
Aceste boli prezintă o defecțiune comună a așa-numitelor cilii (procese mici, antene), care apar la majoritatea celulelor organismului uman. Ciliopatiile se caracterizează prin tranziții curgătoare și suprapuneri între diferite boli ciliare.
Simptome, afectiuni si semne
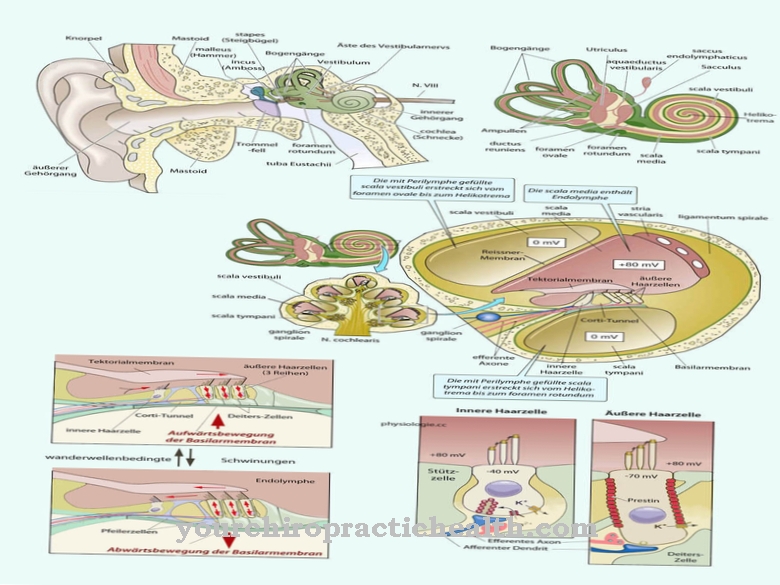
Principala caracteristică a distrofiei retiniene ereditare este un termen generic care descrie debutul pierderii funcției și degenerarea (distrugerea) ulterioară a fotoreceptorilor. Ele duc la o pierdere progresivă (progresivă) a funcției vizuale. Tulburările vizuale care progresează rapid apar de obicei foarte devreme la copii când au între patru și zece ani. Se fac simțite în moduri diferite, în funcție de fotoreceptorii afectați.
Ca o "formă de con de tijă", cu cursul caracteristic al retinitei pigmentare (RP), boala își are originea în periferia retinei (retina exterioară) și se dezvoltă în degenerare maculară (distrugerea vederii ascuțite) printr-o pierdere progresivă a câmpului vizual.
Odată cu obezitatea (obezitatea), corpul prezintă o acumulare patologică de țesut gras. În cazul BBS, acumulările crescute anormal de grăsime pe picioare, stomac, fese, brațe, piept și șolduri apar mai ales ca obezitate a trunchiului, trunchiul, picioarele și coapsele fiind afectate în special. Polidactila este un simptom vizibil și o caracteristică semnificativă a sindromului Bardet-Biedl. Constatarea nu este ușoară, deoarece poliactila rudimentară este corectată chirurgical după naștere.
Razele X sunt capabile să ofere informații suplimentare. Polidactila poate să apară cu semne diferite, de exemplu, ca un picior sau un apendic rudimentar. Un deget sau deget poate fi format suplimentar sau doar parțial. Hexadactila unilaterală de pe picior și / sau mână are o legătură suplimentară, hexadactila bilaterală apare atât la picioare, cât și / sau la mâini.
Degetele de la picioare sau degetele care s-au dezvoltat împreună (sintactic) și scurtarea unuia sau mai mult degetelor de la picioare sau degetelor (brachydactyly) sunt, de asemenea, semne ale BBS. Doar câțiva pacienți au toate extremitățile afectate. Întârzierea dezvoltării mintale este diferită. Doar un număr mic dintre cei afectați prezintă o retardare mentală severă. O inteligență în mod normal este posibilă.
Copiii învață să vorbească și să meargă târziu și, uneori, prezintă probleme de comportament, cum ar fi tulburările de anxietate. Comportamentele compulsive sau autiste, un prag scăzut pentru frustrare și emoționalitatea instabilă sunt alte efecte secundare posibile. Este preferat familiarul, dar modificările sunt respinse. Anomalii în organele genitale interne și externe sunt frecvente.
Alte modificări sunt hipospadiile (deschiderea uretrală este deasupra sau mai jos, în loc de partea din față a penisului), abdomenul sau testiculele inghinale, constricțiile uretrale, constricția preputului și supapele uretrale posterioare. La pacienții de sex feminin, se cunosc atrezie vaginală (vaginul nu este deschis), deschideri uretrale lipsă și labii interioare reduse.
Nu este neobișnuit ca femeile afectate să aibă cicluri menstruale neregulate. Modificările rinichilor sunt reacții adverse frecvente. Constatarea depinde de examinarea tractului urinar inferior și a rinichilor care utilizează ultrasunete (sonografie).
Diagnosticul și cursul bolii
Sindromul Bardet-Biedl (BBS) are șase simptome principale, dar nu apar împreună în fiecare caz. Medicii își asumă o constatare corespunzătoare dacă sunt prezente cel puțin patru dintre simptomele principale. În mod alternativ, există o mare probabilitate ca boala să fie prezentă dacă pacientul prezintă trei simptome principale și două simptome secundare.
Cele șase simptome principale sunt distrofia retiniană, obezitatea (acumularea anormală a țesutului adipos, fiind supraponderal), polidactilie (exces de degetele de la picioare și / sau degetele), retardare mentală (întârziere a dezvoltării mentale), hipogenitalism (organe genitale subdezvoltate) și boli renale. Simptomele secundare de frecvență scăzută includ întârzieri de vorbire, deficiențe de vorbire, malformații cardiace, ataxie (coordonarea mișcării afectate), astm, diabet zaharat (diabet), boala Crohn (inflamația intestinului gros și / sau intestinului subțire), displazie a coastelor și a vertebrelor și cifoscoza (anemie vertebrală) pe.
complicaţiile
Cu sindromul Laurence-Moon-Biedl-Bardet, cei afectați de obicei suferă de o pierdere a funcției vizuale. Pierderea nu se produce brusc, ci treptat. În cel mai rău caz, cei afectați vor rămâne complet orbi, care de obicei nu mai pot fi tratați.
Mai ales la tineri și copii, orbirea poate duce la reclamații psihologice severe sau chiar la depresie. Pacienții sunt limitați clar în viața lor de zi cu zi și suferă de un câmp vizual mult redus. În multe cazuri, sindromul Laurence-Moon-Biedl-Bardet duce, de asemenea, la probleme de comportament, astfel încât, în special, copiii pot suferi de intimidare sau tachinare.
Dezvoltarea copiilor este semnificativ întârziată și restricționată de sindrom. De asemenea, pot apărea tulburări de anxietate. Nu este neobișnuit ca sindromul Laurence-Moon-Biedl-Bardet să conducă la plângeri psihologice și depresie la rude sau părinți. Un tratament cauzal al sindromului Laurence-Moon-Biedl-Bardet nu este posibil din păcate.
Unele reclamații pot fi limitate. Cu toate acestea, nu apare un curs complet pozitiv al bolii. Sindromul nu reduce speranța de viață a pacientului. În unele cazuri, cei afectați au nevoie uneori de ajutor de la alte persoane în viața lor de zi cu zi.
Când trebuie să te duci la doctor?
Deoarece sindromul Laurence-Moon-Biedl-Bardet este o boală ereditară, diagnosticul poate fi făcut în pântec. Cel târziu după naștere, medicul trebuie consultat dacă se observă simptome tipice, cum ar fi tulburări de vedere sau obezitate. Malformațiile degetelor de la picioare și ale degetelor sunt, de asemenea, un indicator clar al unei boli.Părinții care observă simptome la copilul lor trebuie să informeze imediat medicul pediatru.
Un examen cuprinzător oferă informații despre boală. După aceea, terapia este de obicei inițiată direct, care constă în diverse tratamente de către ortopedi, neurologi, oftalmologi, internisti și terapeuți, precum și de către fizioterapeuți. Vizitele suplimentare la medic sunt necesare dacă tratamentul nu are efectul dorit. De asemenea, este necesar un sfat medical în situații de urgență, de exemplu dacă copilul cade ca urmare a unei malformații sau are brusc o convulsie. Dacă bolnavul prezintă semne de disconfort emoțional, părinții trebuie să consulte un terapeut adecvat. Copiii mai în vârstă pot contacta psihologul școlar împreună cu părinții lor și pot discuta despre măsuri adecvate.
Terapie și tratament
Această boală apare pe baza moștenirii recesive autosomale, ceea ce înseamnă că ambele copii (alele) ale unei gene BBS prezintă o schimbare (mutație). Părinții pacientului sunt „cu sânge amestecat” și fiecare poartă o alelă modificată și neschimbată a genei corespunzătoare. Nu au boala. Copiii se îmbolnăvesc numai dacă tatăl și mama lor transmit alele mutate. La copii suplimentari, probabilitatea de repetare este de 25 la sută.
O opțiune de terapie cauzală nu este încă cunoscută, deoarece anumite simptome ale bolii nu pot fi încă atribuite în mod concludent diferitelor modificări genetice. Simptomele și manifestările lor apar diferit chiar și la frații bolnavi. Deoarece imaginea completă caracteristică a BBS este prezentă doar în cazuri rare, în special la copii mici, un diagnostic corespunzător este dificil.
Datorită simptomelor oligosimptomatice frecvente, cu care apar foarte puține simptome atipice și doar ușor pronunțate, alte imagini clinice posibile trebuie luate în considerare în diagnosticul diferențial. Modificările în aceeași genă pot duce la imagini clinice diferite, de exemplu, sindromul Joubert, Bardet-Biedl sau Meckel-Gruber.
Perspective și prognoză
Prognosticul pentru prezența sindromului Laurence-Moon-Biedl-Bardet este, în general, slab, deoarece malformațiile multiple sunt congenitale și incurabile. Dacă apar patru dintre cele șase simptome principale, diagnosticul sindromului Laurence-Moon-Biedl-Bardet este confirmat. La simptomele principale se adaugă numeroase simptome secundare. Aceasta include o orbire înfiorătoare.
Datorită complexității simptomelor, nu există nicio perspectivă de vindecare. Există doar o șansă mediocră de ameliorare a simptomelor vizibile. Numărul de malformații și tulburări posibile în sindromul Laurence-Moon-Biedl-Bardet este atât de mare încât boala ereditară este greu de tratat. În orice caz, cursul acestei boli genetice nu poate fi influențat. Cu toate acestea, simptomele prezente pot fi atenuate parțial.
Cu toate acestea, prognosticul general slab nu reduce speranța de viață a persoanelor afectate. La o vârstă înaintată și după ce orbește, persoanele afectate pot depinde permanent de ajutor sau îngrijire. Prin eforturi medicale interdisciplinare, mulți bolnavi cu sindromul Laurence-Moon-Biedl-Bardet pot experimenta un curs ceva mai ușor al bolii.
Problemele vizuale din ce în ce mai mari reprezintă o parte dificilă de tratat și problematică a bolii.Deficiențele vizuale crescânde apar deja la copiii mici afectați. Se agravează în timp. Problemele de vedere nu trebuie să conducă la orbire la toți cei afectați. Sechelele psihologice ale sindromului Laurence-Moon-Biedl-Bardet pot fi de obicei tratate bine.
profilaxie
Prevenirea în sensul prevenirii acestei boli nu este posibilă. Este importantă monitorizarea regulată a simptomelor și însoțirea simptomelor. Verificările repetate ale tensiunii arteriale și ale funcției renale, sfaturi nutriționale, fizioterapie și terapie ocupațională, precum și logopedie sunt posibile abordări terapeutice.
Dupa ingrijire
În majoritatea cazurilor, persoanele afectate de sindromul Laurence-Moon-Biedl-Bardet nu au la dispoziție opțiuni speciale de urmărire, astfel încât un medic trebuie să fie contactat și consultat foarte devreme în această boală. De regulă, auto-vindecarea nu poate apărea, de aceea tratamentul medicului este întotdeauna necesar.
Deoarece sindromul Laurence-Moon-Biedl-Bardet este o boală ereditară, persoana în cauză ar trebui să facă un examen genetic și un sfat, dacă dorește să aibă copii, astfel încât sindromul Laurence-Moon-Biedl-Bardet să nu treacă la urmașii lor este transmis mai departe. În multe cazuri, cei afectați depind de intervențiile chirurgicale pentru a atenua malformațiile și deformările.
Aici, persoana afectată ar trebui să se odihnească definitiv după procedură și să aibă grijă de corpul său. Efortul sau alte activități fizice și stresante trebuie evitate în orice caz pentru a nu împovăra inutil corpul. Deoarece sindromul Laurence-Moon-Biedl-Bardet poate duce și la un comportament anormal, părinții ar trebui să sprijine și să încurajeze copilul în dezvoltare. Pentru a preveni tulburările psihice sau depresia sunt necesare, de asemenea, discuții amoroase și intense cu copilul.
Puteți face asta singur
Sindromul Laurence-Moon-Biedl-Bardet prezintă simptome diverse, pacientul suferind adesea cel mai mult din cauza funcției vizuale afectate. Chiar și la copii, abilitatea obișnuită de a vedea începe să se deterioreze, astfel încât părinții sunt cei care îi prezintă copilului la medic și astfel grăbesc diagnosticul. În acest fel, boala poate fi tratată rapid, deși până în prezent opțiunile de tratament au fost doar de natură simptomatică.
Tulburările vizuale cresc din ce în ce mai mult la copiii bolnavi și, astfel, afectează considerabil viața de zi cu zi, astfel încât calitatea vieții celor afectați scade. Deoarece problemele de vedere dezvoltă numeroase dificultăți pentru pacient atunci când urmează școala, în timpul liber și în ceea ce privește integritatea fizică a acestuia. Riscul de accidente crește, de asemenea, semnificativ, de exemplu în traficul rutier. Acesta este motivul pentru care părinții își însoțesc copiii bolnavi ori de câte ori este posibil sau angajează personal de asistență medicală, astfel încât pacientul să nu fie lăsat să se arate pentru el însuși.
În unele cazuri, boala se răspândește până la orbire. Întrucât o astfel de dezvoltare este deja evidentă în prealabil, pacienții se pregătesc pentru aceasta. Părinții reproșează spațiul de locuit astfel încât acesta să nu conțină surse de pericol pentru persoana cu deficiențe de vedere. În plus, victimele nevăzătoare învață cum să folosească un băț lung, astfel încât să se poată deplasa independent în afara propriei case.




.jpg)




.jpg)





.jpg)